例如,水:
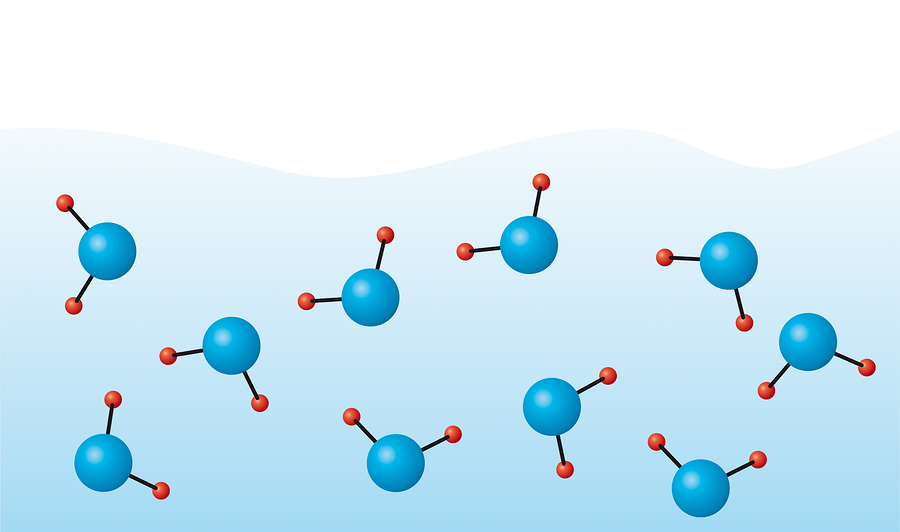
显然,一个分子内的氢和氧与相同的氧与相邻分子的氢发生完全不同的相互作用。因此,区分了分子内和分子间的相互作用。分子间的相互作用可以通过短程和库仑对势来指定,这在先前的文章中已有讨论。在这里,我们将关注分子内。
分子内相互作用的最常见类型是化学键(价键)。化学键是由势能对键合原子之间距离的功能依赖性决定的,也就是说,实际上是由同一对电位决定的。但是,与普通对电位不同,这种相互作用不是针对某些类型的粒子,而是针对特定的一对粒子(通过其指数)。化学键电位最常见的功能形式是谐波电位:
其中r是粒子之间的距离,k是键刚度常数,r 0是平衡键长;和莫尔斯电势:
其中D是势阱的深度,参数α表示势阱的宽度。
分子内相互作用的另一种类型是键角。让我们再次看一下标题图片。为什么分子用一个角表示,因为静电力应该提供氢离子之间的最大距离,该距离对应于等于180°的HOH角?事实是,并非所有内容都绘制在图中。从学校化学课程中,您可以回想起氧气中还有2个单独的电子对,它们之间的相互作用扭曲了角度:

在经典的分子动力学中,通常不引入诸如电子或电子云之类的物体,因此,为了模拟“正确”的角度,使用了键角的电势,即 势能对3个粒子坐标的函数依赖性。最方便的此类电势之一是谐波余弦:
其中θ是由粒子三重态形成的角度,k是刚度常数,θ0是平衡角。
存在更高阶的分子内电势,例如扭转角,但它们比键角更为人为。
在具有预定折射率的粒子之间添加相互作用是微不足道的。对于链接,我们存储一个数组,其中包含链接的粒子的索引和交互类型。我们为每个线程提供自己的链接范围进行处理,您已完成。同样对于键角。因此,我们将立即为自己完成任务:增加创建/删除化学键和运行时键角的功能。这立即使我们脱离了经典分子动力学的范畴,开辟了可能性的新视野。否则,您可以简单地从现有软件包中下载某些内容,例如LAMMPS,DL_POLY或GROMACS,尤其是因为它们是免费分发的。
现在获取一些代码。让我们在主结构中添加适当的字段:
//bonds:
int nBond; // number of bonds
int mxBond; // maximal number of bonds
int4* bonds; // array of bonds
int* nbonds; // count of bond for a given atom
int* neighToBind; // a neighbor of a given atom for binding
int* canBind; // flags that atom[iat] can be bind
int* r2Min; // distances for the nearest neighbor (used for binding)
int* parents; // indexes of one of the atom bonded with a given
cudaBond* bondTypes;
int** def_bonds; // array[nSpec][nSpec] of default bond types
int** bindBonds; // array[nSpec][nSpec] bond types created by binding
float** bindR2; // square of binding distance [nSpec][nSpec]
//angles:
int nAngle; // number of angles
int mxAngle;
int4* angles; // array of angles
int* nangles; // number of angles for given atom
int* oldTypes;
cudaAngle* angleTypes;
int* specAngles; // [nSp] angle type formed by given species
链接和角度的数量是可变的,但是几乎总是可以估计可能的最大值,并立即在最大值下分配内存,以免过度分配内存,nBond和mxBond字段分别表示当前的链接数量和最大值。键数组将包含要键合的原子的索引,键的类型和键创建的时间(如果我们突然对平均键寿命这样的统计数据感兴趣)。角度阵列将保存形成键角和键角类型的原子三重态的索引。的bondTypes和angleTypes阵列将包含可能键电位和角度的特性。它们的结构如下:
struct cudaBond
{
int type; // potential type
int spec1, spec2; // type of atoms that connected by this bond type
int new_type[2]; // bond type after mutation
int new_spec1[2], new_spec2[2];
int mxEx, mnEx; // flags: maximum or minimum of bond length exists
float p0, p1, p2, p3, p4; // potential parameters
float r2min, r2max; // square of minimal and maximal bond length
float (*force_eng)(float r2, float r, float &eng, cudaBond *bond); // return energy
int count; // quantity of such bonds
float rSumm; // summ of lentghs (for mean length calculation)
int rCount; // number of measured lengths (for mean length calculation)
int ltSumm, ltCount; // for calculation of lifetime
};
struct cudaAngle
{
int type; // potential type
float p0, p1, p2; // potential parameters
void (*force_eng)(int4* angle, cudaAngle* type, cudaMD* md, float& eng);
};
type 字段定义电势的功能形式,new_type,new_spec1和new_spec是键类型的索引以及在键更改(断开或转变为其他类型的键)后要键合的原子类型。这些字段表示为具有两个元素的数组。第一种对应于长度小于r2min 1/2的情况,第二种对应于长度超过r2max 1/2的情况...该算法最困难的部分是应用所有键的特性,同时考虑到其断裂和转化的可能性,以及其他流动可能破坏相邻键的事实,这导致了键合原子类型的改变。让我用同一水的例子来解释。最初,该分子是电中性的,化学键是由氢和氧共有的电子形成的。粗略地说,我们可以说氢和氧原子上的电荷为零(实际上,电子密度移到了氧上,因此氢,δ+和氧-2δ-上有一个小的加号)。如果我们打破键,氧将最终为自己带来一个电子,而氢将释放它。得到的颗粒为H +和O - 。总共获得了5种类型的粒子,让我们按常规指定它们:H,H +,O,O-,O 2-。如果我们将两个氢都从水分子中分离出来,就会形成后者。因此,反应:
H ^ 2 -O - >ħ + + OH -
和
OH - - > H ^ + + O 2-。
化学专家会纠正我的观点,即在水的标准条件下,分解的第一阶段实际上没有进行(平衡时,只有10 7中有一个分子分解成离子,甚至还不如所写)。但是对于算法的描述,这样的方案将是说明性的。假设一个流处理水分子中的一个键,而另一个流处理同一分子中的第二个键。碰巧这两个关系都需要打破。然后一个流应变换原子成H +和O - ,并且所述第二成H +和O 2-。但是,如果数据流同时做到这一点,在程序开始的时候,氧气是在O状态和两个流转换制成O - ,这是不正确。我们需要以某种方式防止这种情况。处理化学键的功能的框图:

我们检查原子的当前类型是否对应于连接的类型,如果不是,则从默认类型的表中获取(必须预先编译),然后确定原子之间的距离的平方(r 2),如果连接隐含着最大或最小长度,则检查它是否不存在我们是否超越了这些界限。如果这样做,那么我们需要更改连接的类型或将其删除,并且在两种情况下都需要更改原子的类型。为此,将使用atomicCAS功能-我们将当前原子类型与应有的原子类型进行比较,在这种情况下,我们将其替换为新的原子类型。如果原子的类型已经被另一个线程更改,则返回到开头以覆盖链接类型。最坏的情况是如果我们设法更改第一个原子的类型,但不能更改第二个原子的类型。现在返回已经太晚了,因为在我们更改了第一个原子之后,其他线程已经可以对其进行处理。出路是什么?我建议我们假装我们正在破坏/更改其他类型的连接,而不是开始时使用的那种。我们发现最初的第一个原子与更改的第二个原子之间应该建立什么样的连接,并根据与最初预期相同的规则对其进行处理。如果在这种情况下原子的类型再次更改,我们将再次使用相同的方案。这里暗含着一种新型的键具有相同的特性-它以相同的长度断裂等,并且在断裂过程中形成的颗粒是按需的。由于用户触摸了此信息,因此我们将责任从我们的程序转移到他身上,他必须正确设置所有内容。代码:
__global__ void apply_bonds(int iStep, int bndPerBlock, int bndPerThread, cudaMD* md)
{
int def;
int id1, id2; // atom indexes
int old, old_spec2, spec1, spec2, new_spec1, new_spec2; // atom types
int new_bond_type;
int save_lt, need_r, loop; // flags to save lifetime, to need to calculate r^2 and to be in ‘while’ loop
int mnmx; // flag minimum or maximum
int action; // flag: 0 - do nothing, 1 - delete bond, 2 - transform bond
cudaBond *old_bnd, *cur_bnd; // old bond type, current bond type
float dx, dy, dz, r2, r;
float f, eng = 0.0f;
__shared__ float shEng;
#ifdef DEBUG_MODE
int cnt; // count of change spec2 loops
#endif
if (threadIdx.x == 0)
{
shEng = 0.0f;
}
__syncthreads();
int id0 = blockIdx.x * bndPerBlock + threadIdx.x * bndPerThread;
int N = min(id0 + bndPerThread, md->nBond);
int iBnd;
for (iBnd = id0; iBnd < N; iBnd++)
if (md->bonds[iBnd].z) // the bond is not broken
{
// atom indexes
id1 = md->bonds[iBnd].x;
id2 = md->bonds[iBnd].y;
// atom types
spec1 = md->types[id1];
spec2 = md->types[id2];
old_bnd = &(md->bondTypes[md->bonds[iBnd].z]);
cur_bnd = old_bnd;
save_lt = 0;
need_r = 1;
loop = 1;
#ifdef DEBUG_MODE
cnt = 0;
#endif
if ((cur_bnd->spec1 == spec1)&&(cur_bnd->spec2 == spec2))
{
//ok
}
else
if ((cur_bnd->spec1 == spec2) && (cur_bnd->spec2 == spec1))
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
//... then ok
}
else // atom types do not correspond to bond types
{
save_lt = 1;
}
// end initial stage
while (loop)
{
if (save_lt)
{
def = md->def_bonds[spec1][spec2];
if (def == 0) // these atom types do not form a bond
{
#ifdef DEBUG_MODE
printf("probably, something goes wrong\n");
#endif
action = 1; // delete
break;
}
else
{
//! change bond type and go on
if (def < 0)
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
def = -def;
}
md->bonds[iBnd].z = def;
cur_bnd = &(md->bondTypes[def]);
}
} // end if (save_lt)
// calculate distance (only once)
if (need_r)
{
dx = md->xyz[id1].x - md->xyz[id2].x;
dy = md->xyz[id1].y - md->xyz[id2].y;
dz = md->xyz[id1].z - md->xyz[id2].z;
delta_periodic(dx, dy, dz, md);
r2 = dx * dx + dy * dy + dz * dz;
need_r = 0;
}
action = 0; // 0 - just cultivate bond 1 - delete bond 2 - transform bond
if ((cur_bnd->mxEx) && (r2 > cur_bnd->r2max))
{
mnmx = 1;
if (cur_bnd->new_type[mnmx] == 0) // delete bond
action = 1;
else
action = 2; // modify bond
}
else if ((cur_bnd->mnEx) && (r2 < cur_bnd->r2min))
{
mnmx = 0;
action = 2; // at minimum only bond modification possible
}
// end select action
// try to change atom types (if needed)
if (action)
{
save_lt = 1;
new_spec1 = cur_bnd->new_spec1[mnmx];
new_spec2 = cur_bnd->new_spec2[mnmx];
//the first atom
old = atomicCAS(&(md->types[id1]), spec1, new_spec1);
if (old != spec1)
{
spec1 = old;
spec2 = md->types[id2]; // refresh type of the 2nd atom
// return to begin of the ‘while’ loop
}
else // types[id1] have been changed
{
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
#endif
old_spec2 = spec2;
while ((old = atomicCAS(&(md->types[id2]), old_spec2, new_spec2)) != old_spec2)
{
//! the worst variant: this thread changes atom 1, other thread changes atom 2
// imagine that we had A-old bond with the same behavior
def = md->def_bonds[spec1][old];
#ifdef DEBUG_MODE
if (def == 0)
{
printf("UBEH[001]: in apply_bonds, change atom types. There are no bond types between Species[%d] and [%d]\n", spec1, old);
break;
}
#endif
if (def < 0) // spec1 -> new_spec2 spec2 -> newSpec1
{
cur_bnd = &(md->bondTypes[-def]);
new_spec2 = cur_bnd->new_spec1[mnmx];
}
else // direct order
{
cur_bnd = &(md->bondTypes[def]);
new_spec2 = cur_bnd->new_spec2[mnmx];
}
old_spec2 = old;
#ifdef DEBUG_MODE
cnt++;
if (cnt > 10)
{
printf("UBEH[002]: too many atempst to change spec2 = %d\n", spec2);
break;
}
#endif
}
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
loop = 0;
}
//end change types
} // end if (action)
else
loop = 0; // action == 0, out of cycle
} // end while(loop)
if (action == 2)
{
new_bond_type = cur_bnd->new_type[mnmx];
if (new_bond_type < 0)
{
md->bonds[iBnd].x = id2;
md->bonds[iBnd].y = id1;
new_bond_type = -new_bond_type;
}
md->bonds[iBnd].z = new_bond_type;
cur_bnd = &(md->bondTypes[new_bond_type]);
}
// perform calculations and save mean bond length
if (action != 1) // not delete
{
r = sqrt(r2);
f = cur_bnd->force_eng(r2, r, eng, cur_bnd);
atomicAdd(&(md->frs[id1].x), f * dx);
atomicAdd(&(md->frs[id2].x), -f * dx);
atomicAdd(&(md->frs[id1].y), f * dy);
atomicAdd(&(md->frs[id2].y), -f * dy);
atomicAdd(&(md->frs[id1].z), f * dz);
atomicAdd(&(md->frs[id2].z), -f * dz);
atomicAdd(&(cur_bnd->rSumm), r);
atomicAdd(&(cur_bnd->rCount), 1);
}
else //delete bond
{
// decrease the number of bonds for atoms
atomicSub(&(md->nbonds[id1]), 1);
atomicSub(&(md->nbonds[id2]), 1);
md->bonds[iBnd].z = 0;
// change parents
exclude_parents(id1, id2, md);
}
if (save_lt)
{
keep_bndlifetime(iStep, &(md->bonds[iBnd]), old_bnd);
if (action != 1) // not delete
atomicAdd(&(cur_bnd->count), 1);
atomicSub(&(old_bnd->count), 1);
}
} // end main loop
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engBond), shEng);
}
在代码中,我使用了预处理程序指令来检查由于用户疏忽而引起的情况。您可以关闭它们以提高性能。该函数实现了上述方案,但又包裹在另一个循环中,该循环遍历此线程负责的链接范围。在下文中,键类型的标识符可以为负,这意味着键中原子的顺序必须颠倒(例如,OH和HO键是同一键,但是在算法中,顺序很重要,以表明这一点我使用了相反的索引符号),因此invert_bond函数使其描述起来过于琐碎。Delta_periodic函数应用周期性边界条件来协调差异。如果我们不仅需要更改键,还需要更改键角(USE_NEWANG指令),那么我们需要标记已更改类型的原子(稍后将对此进行详细介绍)。为了排除相同原子与键的重新结合,母体数组存储与数据关联的原子之一的索引(此安全网并非在所有情况下都起作用,但对于我来说就足够了)。如果断开某种连接,则需要从parents数组中删除相应的原子索引,这是通过exclude_parents函数完成的:
__device__ void exclude_parents(int id1, int id2, cudaMD* md)
// exclude id1 and id2 from parents of each other (if they are)
// and seek other parents if able
{
// flags to clear parent
int clear_1 = 0;
int clear_2 = 0;
int i, flag;
if (md->parents[id1] == id2)
clear_1 = 1;
if (md->parents[id2] == id1)
clear_2 = 1;
i = 0;
while ((i < md->nBond) && (clear_1 || clear_2))
{
if (md->bonds[i].z != 0)
{
flag = 0;
if (clear_1)
{
if (md->bonds[i].x == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_1 = 0;
i++;
continue;
}
}
if (clear_2)
{
if (md->bonds[i].x == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_2 = 0;
i++;
continue;
}
}
}
i++;
}
// be on the safe side
if (clear_1)
md->parents[id1] = -1;
if (clear_2)
md->parents[id2] = -1;
}
不幸的是,该功能贯穿整个链接数组。我们学习了如何处理和删除链接,现在我们需要学习如何创建链接。以下功能标记了适合于形成化学键的原子:
__device__ void try_to_bind(float r2, int id1, int id2, int spec1, int spec2, cudaMD *md)
{
int r2Int; // (int)r2 * const
// save parents to exclude re-linking
if (md->parents[id1] == id2)
return;
if (md->parents[id2] == id1)
return;
if (md->bindBonds[spec1][spec2] != 0)
{
if (r2 < md->bindR2[spec1][spec2])
{
r2Int = (int)(r2 * 100);
if (atomicMin(&(md->r2Min[id1]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id1] = id2 + 1; // as 0 is reserved for no neighbour
md->canBind[id1] = 1;
}
// similar for the second atom
if (atomicMin(&(md->r2Min[id2]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id2] = id1 + 1; // as 0 is reserved for no bind
md->canBind[id2] = 1;
}
}
}
}
bindBonds 矩阵存储有关这些类型的原子是否可以形成键以及是否可以形成键的信息。bindR2矩阵存储绑定所需原子之间的最大距离。如果所有条件都符合条件,则我们检查其他邻居的原子是否适合于键合,但更接近键合。
有关到邻居的最近距离的信息存储在r2Min数组中(为方便起见,该数组为int类型,并且将其值乘以常数100转换为该值)。如果检测到的邻居是最接近的邻居,那么我们会记住其在neighToBind数组中的索引并设置canBind标志...确实存在危险,当我们继续更新索引时,另一个线程已覆盖了最小值,但这并不是那么关键。建议在遍历第一部分中描述的成对原子的函数(例如cell_list或all_pair)中调用此函数。绑定本身:
__global__ void create_bonds(int iStep, int atPerBlock, int atPerThread, cudaMD* md)
// connect atoms which are selected to form bonds
{
int id1, id2, nei; // neighbour index
int btype, bind; // bond type index and bond index
cudaBond* bnd;
int spec1, spec2; // species indexes
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
{
nei = md->neighToBind[iat];
if (nei) // neighbour exists
{
nei--; // (nei = spec_index + 1)
if (iat < nei)
{
id1 = iat;
id2 = nei;
}
else
{
id1 = nei;
id2 = iat;
}
// try to lock the first atom
if (atomicCAS(&(md->canBind[id1]), 1, 0) == 0) // the atom is already used
continue;
// try to lock the second atom
if (atomicCAS(&(md->canBind[id2]), 1, 0) == 0) // the atom is already used
{
// unlock the first one back
atomicExch(&(md->canBind[id1]), 1);
continue;
}
// create bond iat-nei
bind = atomicAdd(&(md->nBond), 1);
#ifdef DEBUG_MODE
if (bind >= md->mxBond)
{
printf("UBEH[003]: Exceed maximal number of bonds, %d\n", md->mxBond);
}
#endif
spec1 = md->types[id1];
spec2 = md->types[id2];
#ifdef USE_NEWANG // save that we have changed atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
btype = md->bindBonds[spec1][spec2];
if (btype < 0)
{
// invert atoms order
md->bonds[bind].x = id2;
md->bonds[bind].y = id1;
md->bonds[bind].z = -btype;
bnd = &(md->bondTypes[-btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec2;
md->types[id2] = bnd->spec1;
}
else
{
md->bonds[bind].x = id1;
md->bonds[bind].y = id2;
md->bonds[bind].z = btype;
bnd = &(md->bondTypes[btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec1;
md->types[id2] = bnd->spec2;
}
atomicAdd((&bnd->count), 1);
md->bonds[bind].w = iStep; // keep time of the bond creation for lifetime calculation
atomicAdd(&(md->nbonds[id1]), 1);
atomicAdd(&(md->nbonds[id2]), 1);
// replace parents if none:
atomicCAS(&(md->parents[id1]), -1, id2);
atomicCAS(&(md->parents[id2]), -1, id1);
}
}
// end loop by atoms
}
该函数将阻塞一个原子,然后阻塞第二个原子,如果成功阻塞了两个原子,则会在它们之间建立连接。在该函数的最开始,对原子索引进行排序,以排除一个线程阻塞一对中的第一个原子,而另一个线程阻塞同一对中的第二个原子的情况,两个线程都成功通过了第一个检查而第二个则失败,因此两个线程都没有通过连接不会创建它们。最后,我们需要删除在apply_bonds函数中标记为删除的链接:
__global__ void clear_bonds(cudaMD* md)
// clear bonds with .z == 0
{
int i = 0;
int j = md->nBond - 1;
while (i < j)
{
while ((md->bonds[j].z == 0) && (j > i))
j--;
while ((md->bonds[i].z != 0) && (i < j))
i++;
if (i < j)
{
md->bonds[i] = md->bonds[j];
md->bonds[j].z = 0;
i++;
j--;
}
}
if ((i == j) && (md->bonds[i].z == 0))
md->nBond = j;
else
md->nBond = j + 1;
}
我们只需将“空化”链接移动到数组的末尾,并减少实际的链接数。不幸的是,代码是串行的,但是我不确定将其并行化是否会带来明显的影响。由cudaBond结构的force_eng字段指示的用于计算原子上的实际结合能和力的函数仍然省略,但是它们与第一部分中所述的成对电势函数完全相似。
价角
对于价键,我将介绍一些假设,以使算法和功能更轻松,结果,它们将比价键更简单。首先,键角的参数应取决于所有三个原子,但是在这里我们将假设键角的类型仅决定原子在其顶点处的位置。我提出了最简单的形成/去除角的算法:每当我们改变原子的类型时,我们都会在相应的数组oldTypes []中记住这一事实。数组的大小等于原子数,最初用-1填充。如果函数更改原子的类型,则将-1替换为原始类型的索引。对于所有已更改类型的原子,请删除所有键角,并遍历此原子的所有键以添加相应的角:
__global__ void refresh_angles(int iStep, int atPerBlock, int atPerThread, cudaMD *md)
// delete old angles and create new ones for atoms which change their type
{
int i, j, n, t, ang;
int nei[8]; // bonded neighbors of given atom
int cnt;
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
if (md->oldTypes[iat] != -1)
{
i = 0;
n = md->nangles[iat];
while (n && (i < md->nAngle))
{
if (md->angles[i].w)
if (md->angles[i].x == iat)
{
n--;
md->angles[i].w = 0;
}
i++;
}
// create new angles
t = md->specAngles[md->types[iat]]; // get type of angle, which formed by current atom type
if (t && (md->nbonds[iat] > 1)) // atom type supports angle creating and number of bonds is enough
{
// search of neighbors by bonds
i = 0; cnt = 0;
n = md->nbonds[iat];
while (n && (i < md->nBond))
{
if (md->bonds[i].z) // if bond isn't deleted
{
if (md->bonds[i].x == iat)
{
nei[cnt] = md->bonds[i].y;
cnt++;
n--;
}
else if (md->bonds[i].y == iat)
{
nei[cnt] = md->bonds[i].x;
cnt++;
n--;
}
}
i++;
}
// add new angles based on found neighbors:
for (i = 0; i < cnt-1; i++)
for (j = i + 1; j < cnt; j++)
{
ang = atomicAdd(&(md->nAngle), 1);
md->angles[ang].x = iat;
md->angles[ang].y = nei[i];
md->angles[ang].z = nei[j];
md->angles[ang].w = t;
}
n = (cnt * (cnt - 1)) / 2;
}
md->nangles[iat] = n;
// reset flag
md->oldTypes[iat] = -1;
}
}
specAngles数组包含对应于给定原子类型的键角标识符。以下函数调用所有角度的能量和力的计算:
__global__ void apply_angles(int iStep, int angPerBlock, int angPerThread, cudaMD* md)
// apply valence angle potentials
{
cudaAngle* ang;
// energies of angle potential
float eng;
__shared__ float shEng;
if (threadIdx.x == 0)
shEng = 0.0f;
__syncthreads();
int id0 = blockIdx.x * angPerBlock + threadIdx.x * angPerThread;
int N = min(id0 + angPerThread, md->nAngle);
int i;
for (i = id0; i < N; i++)
if (md->angles[i].w)
{
ang = &(md->angleTypes[md->angles[i].w]);
ang->force_eng(&(md->angles[i]), ang, md, eng);
}
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engAngl), shEng);
}好吧,例如,这些角度的电势给出了谐波余弦函数,这可能表明场force_eng结构cudaAngle:
__device__ void angle_hcos(int4* angle, cudaAngle* type, cudaMD* md, float& eng)
// harmonic cosine valent angle potential:
// U = k / 2 * (cos(th)-cos(th0))^
{
float k = type->p0;
float cos0 = type->p1;
// indexes of central atom and ligands:
int c = angle->x;
int l1 = angle->y;
int l2 = angle->z;
// vector ij
float xij = md->xyz[l1].x - md->xyz[c].x;
float yij = md->xyz[l1].y - md->xyz[c].y;
float zij = md->xyz[l1].z - md->xyz[c].z;
delta_periodic(xij, yij, zij, md);
float r2ij = xij * xij + yij * yij + zij * zij;
float rij = sqrt(r2ij);
// vector ik
float xik = md->xyz[l2].x - md->xyz[c].x;
float yik = md->xyz[l2].y - md->xyz[c].y;
float zik = md->xyz[l2].z - md->xyz[c].z;
delta_periodic(xik, yik, zik, md);
float r2ik = xik * xik + yik * yik + zik * zik;
float rik = sqrt(r2ik);
float cos_th = (xij * xik + yij * yik + zij * zik) / rij / rik;
float dCos = cos_th - cos0; // delta cosinus
float c1 = -k * dCos;
float c2 = 1.0 / rij / rik;
atomicAdd(&(md->frs[c].x), -c1 * (xik * c2 + xij * c2 - cos_th * (xij / r2ij + xik / r2ik)));
atomicAdd(&(md->frs[c].y), -c1 * (yik * c2 + yij * c2 - cos_th * (yij / r2ij + yik / r2ik)));
atomicAdd(&(md->frs[c].z), -c1 * (zik * c2 + zij * c2 - cos_th * (zij / r2ij + zik / r2ik)));
atomicAdd(&(md->frs[l1].x), c1 * (xik * c2 - cos_th * xij / r2ij));
atomicAdd(&(md->frs[l1].y), c1 * (yik * c2 - cos_th * yij / r2ij));
atomicAdd(&(md->frs[l1].z), c1 * (zik * c2 - cos_th * zij / r2ij));
atomicAdd(&(md->frs[l2].x), c1 * (xij * c2 - cos_th * xik / r2ik));
atomicAdd(&(md->frs[l2].y), c1 * (yij * c2 - cos_th * yik / r2ik));
atomicAdd(&(md->frs[l2].z), c1 * (zij * c2 - cos_th * zik / r2ik));
eng += 0.5 * k * dCos * dCos;
}
我不会提供用于消除“变空”的角的功能,它与clear_bonds根本没有区别。
示例
在不假装准确的情况下,我试图描绘单个离子中水分子的组装。成对的势能以白金汉势的形式任意设置,然后增加了以谐波势形式形成键的能力,平衡距离等于水中HO键的长度,为0.96Å。另外,当第二质子与氧结合时,与氧中的顶点的结合角增加。经过10万步之后,第一个分子从随机散布的离子中出现。该图显示了初始(左侧)和最终(右侧)配置:

您可以像这样进行实验:首先让所有原子都相同,但是当它们彼此相邻时,它们会形成键。让结合的原子与游离原子或与另一个类似的结合分子形成另一个键。结果,我们得到了某种自组织,其中原子按链排列:

最后评论
- 在这里,我们仅使用一个约束标准-距离,尽管原则上可能存在其他标准,例如系统的能量。实际上,通常当形成化学键时,能量以热的形式释放。这里没有考虑到这一点,但是您可以尝试实现它,例如,更改粒子速度。
- 粒子之间通过化学键电势进行的相互作用并不能否认粒子仍可以通过分子间对电势和库仑相互作用进行相互作用的事实。当然,可能不计算束缚原子的分子间相互作用,但是在一般情况下,这需要长时间检查。因此,更容易以这样的方式设置化学键电势,使得其与其他电势之和提供期望的功能。
- 由于粒子相互竞争,因此并行执行粒子绑定不仅可以提高速度,而且看起来更加逼真。
好吧,Habré上有几个与这个项目非常接近的项目: